Zygosity check in transgenic animals
This page introduces our method for genotyping (zygosity check) of transgenic mice (Noguchi et al., 2004). Please refer to this reference for detailed explanation.
The genotyping is quite important for proper maintenance of transgenic mouse lines. The method described here was originally developed for creating zygosity check systems of transgenes using PCR with flanking primers. This method is based on genomic sequences flanking the transgene, which has to be first determined by genomic walking technique, the flanking genomic sequences can be also used for chromosomal mapping of the transgene integration site in combination with genome database search. In addition, transgene integration patterns are also clarified by this method in some cases (Suzuki et al., 2006). Therefore, we believe that our method can be used for a wide variety of transgenic research.
Any questions, any suggestions, and any improvements are always welcome. Please contact us if you have any of them.
Methods for zygosity checks in transgenic mice
Many methods have been reported for zyogsity checks of transgenes in transgenic mice. For instance, a famous book on transgenic mouse production, 'Manipulating the mouse embryos (3rd ed.)', describes various methods to identify homozygous transgenic mice as described below.
- Quantification of transgene dosage
- Southern blot analysis
- Quantitative Realtime-PCR
- Quantification transgene products or phenotype (e.g., enzyme activity)
- FISH analysis
- Test breeding
- PCR analysis with flanking primers
Genotyping (zygosity check) methods should be simple, safe, and secure because the genotyping should be routinely performed for maintenance and use of transgenic mice. Southern blots or FISH analyses for all transgenic mice are performed for all mice used would be demanding and time-consuming. It is not easy to prepare DNA samples at preferable concentration every time in order to differentiate clearly homozygous and hemizygous mice by quantitative realtime-PCR. Qualitative analysis (e.g., presence or absence of certain bands) is easier to be performed than quantitative analysis (e.g. same or twice as much PCR products). Qualitative analysis with PCR can be done even though DNA concentrations of sample templates are roughly adjusted. In this sense, the 'PCR with flanking primers' would be most practical for routine zygosity tests of transgenic mice. When we use or maintain transgenic mice, we have to monitor the transgenic status of the mice every time by PCR or some other methods. It would be helpful if you can judge zygosity in addition to the presence of transgenes by doing a simple PCR. But, the problem is how to design flanking primers. In general, we can not predict the chromosomal position where the transgenes integrates nor get sequence information flanking to the transgenes in advance. Therefore, we should determine the flanking genome sequence by some means first, then design flanking primers. Many people think this is not a simple task, but in reality, it is not so difficult. With our methods described below, we determined flanking genome sequence in many cases. Once we got flanking sequences, we can set up a site-specific zygosity check system (we can identify transgenic lines even with the same transgene constructs because the integration sites are not the same among transgenic lines in most cases) and it is easily performed in routine PCR tests. Do you think it would be much easier to do Southern blots to determine zygosity every time ?
Genotyping using PCR with flanking primers
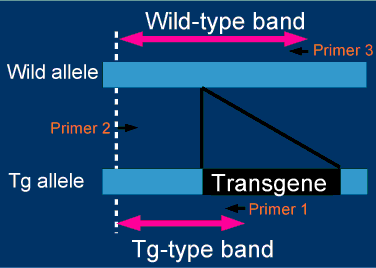
Flanking primers for a zygosity check are designed on the basis of the genomic sequences at both
ends of the integration sites. Right figure summarizes the strategy for zygosity check.
Zygosity is judged by the differential bands produced with flanking primers, which are designed
to distinguish between wild-type(Wild) and transgenic (TG) alleles by amplicon length.
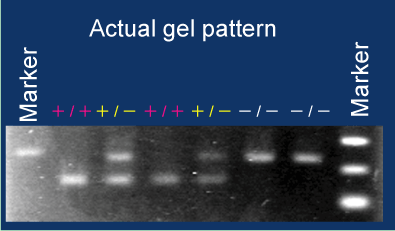
An example of actual zygosity check is shown in the right figure.
Zygosity of six mice was determined by the differential bands on a gel.
In this case, primers are designed so that long and short bands are derived from wild (-) and
Tg (+) alleles, respectively. Zygosity can be clearly judged by the combination of amplicons.
The point is that the PCR amplification should always produce at least one band. No amplicon production indicates inappropriate PCR. It would be a problem if you set up a system that the absence of transgenes is judged only by no amplicon production because this system cannot eliminate a possibility that the PCR itself has a problem. With this system, all samples would be judged as negative when we fail to put DNA templates into PCR tubes (it would be a practical problem when we have to deal with many samples at the same time). For this reason, our method is designed to produce either transgenic-derived, wild-derived, or both bands. If no bands are amplified by PCR, you should re-test the sample.
How to design flanking primers
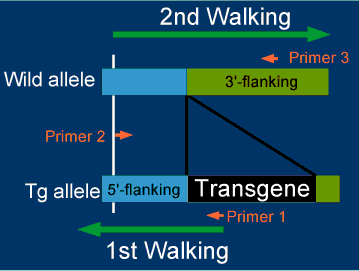
Genomic sequences flanking transgenes are determined by two steps of genomic walkings as shown below. The genomic walking will be explained below.
Procedure 1) 1st Walking (Start from Primer 1)
Determine 5'-flanking sequence using genomic DNA from transgenic mice and design Primer 2.
Procedure 2) 2nd Walking (Start from Primer 2)
Determine 3'-flanking sequence using genomic DNA from non-transgenic, parental strain of mice. In some cases, we can retrieve 3'-flanking sequences from genome databases, instead of doing the 2nd genomic walking.
Procedure 3) Design flanking primers
Design primer 3 based on the 3'-flanking sequence.
Determination of flanking genome sequence by genomic walking
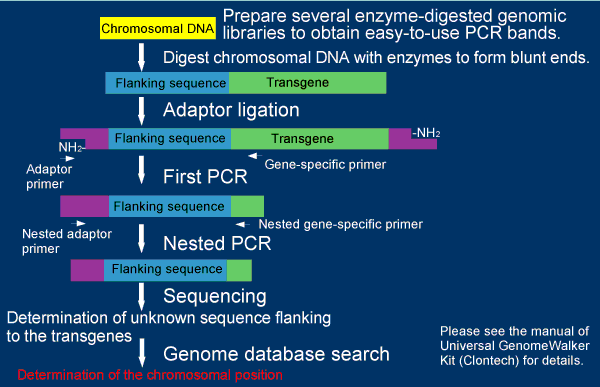
The right figure summarizes the procedure of the genomic walking in this method using the Universal GenomeWalker kit from Clontech with a slight modification (This figure is modified from a chart in a manual of the kit). Six enzyme-digested, adaptor-ligated genomic libraries are constructed from transgenic mice. Fragments flanking the transgenes are amplified by two consecutive PCR amplifications with adaptor and transgene-specific primers. Genomic sequences flanking the transgene are determined by sequencing the fragments. Integration sites of the transgenes were determined by genome database searches.
For practical analyses, you should consider the followings to eliminate false positive amplicons:
- Employ hot-start PCR using so-called hot-start polymerases (chemically-modified would be preferred).
- Perform shuttle PCR (two-step PCR) with high annealing temperature.
- Use longer primers (27-29-mer) to make annealing temperature higher.
- Perform Nested PCR to confirm bands are amplified by two sets of primers
We cannot say we can always determine the flanking genome sequence with this method, but
the odds are not so low. According to our experience, we got sequences in four out of five trails.
An example
Here we show one of our results we actually performed so far.
Transgene construct based on pCAGGS vectors
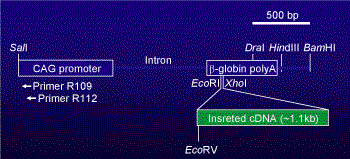
In our laboratory, we usually construct transgenes based on pCAGGS vectors (Niwa et al., 1991). The pCAGGS vector contains CAG-promoter (Cytomegalovirus enhancer + chicken β-actin promoter) and rabbit β-globin polyadenylation sites. If you put your desired DNA fragments into the multicloning site of the vector, you can expect high and systemic expression of the transgene in transgenic mice.
For genomic walking from the transgene based on the pCAGGS vector,
a CMV-enhancer region of the vector would be a better place for primer annealing sites.
The primers for the region would be more specific than the other locations in the vector
because non-mammalian sequence shows low homology to mammalian genome sequences.
We usually use R109 and R112 for the primers to capture the region (See Noguchi et al., 2004 for
sequence info).
Determination of flanking sequence by genomic walking
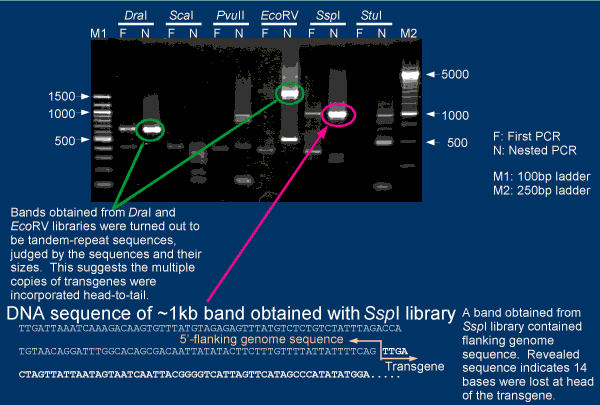
An example of genomic walking using 4C30 mice are shown in the figure.
Ideally, bands which appear in both first and nested PCR's should be sequenced for detecting flanking
sequence of the transgene. But, you may have to adjust PCR conditions for detection of PCR bands.
Very intense PCR bands are often came from tandem repeats of transgenes (e.g., bands in Lanes of DraI and
EcoRV). Such bands are not informative to get flanking sequences.
In the case of 4C30 mice, flanking sequcne of the transgene was determined
by sequensing a band from SspI library.
Chromosomal mapping using Ensembl database
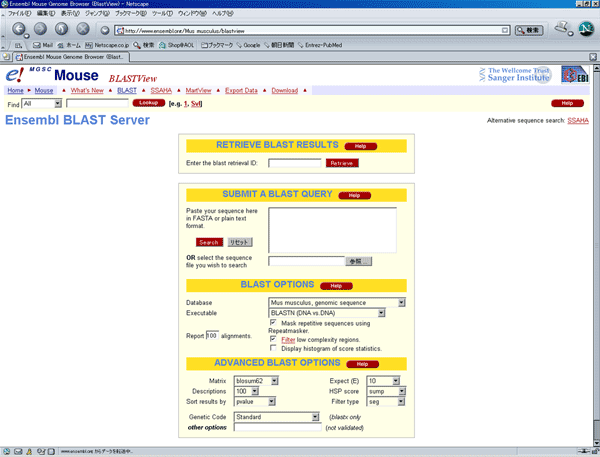
Based on the flanking sequence, the position in the mouse genome was located by BLAST search in the Ensembl database.
The screen image shown above is at the time when the analysis of 4C30 mice was done.
It has been changed into a new version already.
The database is updating (Version 38 in April, 2006),
so you should record which version of database is used for actual analysis.
Example for chromosomal mapping
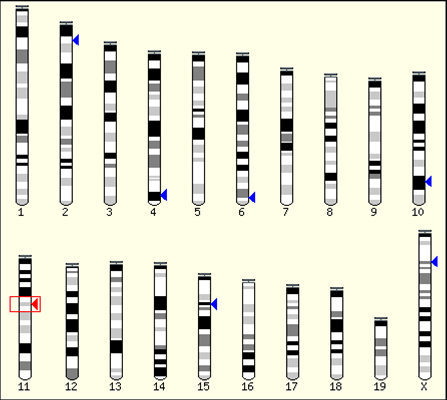
The figure shows the position of flanking sequence of transgene in 4C30 mice, which revealed by the
Ensembl database search. As shown in the figure, the flanking sequence of the transgene, i.e.,
insertion site of the transgene is located in the 11th chromosome. The Ensemble database also tell us
about the additional information around the position of the sequence.
Since sequences around the insertion position can be retrived from the database, we can design flanking primers for zyogosity check
with such sequence information as shown above.
The insertion pattern of the transgene is not so simple in many case, you should consider this for
design of primers. For detailed discussion regarding this problem, please refer to Noguchi et al, 2004.
References
- Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108(2):193-199. [PMID: 1660837]
- Noguchi A, Takekawa N, Einarsdottir T, Koura M, Noguchi Y, Takano K, Yamamoto Y, Matsuda J, Suzuki O. 2004. Chromosomal mapping and zygosity check of transgenes based on flanking genome sequences determined by genomic walking. Exp Anim 53(2):103-111.
- Suzuki O, Hata T, Takekawa N, Koura M, Takano K, Yamamoto Y, Noguchi Y, Uchio-Yamada K, Matsuda J. 2006. Transgene insertion pattern analysis using genomic walking in a transgenic mouse line. Exp. Anim. 55(1):65-69.